杨宝峰院士团队最新成果 | 云序助力揭示RNA修饰m7G调控心肌肥厚的机制研究
导语
心肌肥厚最初是心肌对各种心脏疾病引起的机械性应激的代偿反应。然而,由持续应激引起的病理性心脏肥大以心脏结构和功能改变为特征,是发展为心力衰竭(HF)的危险因素。心肌肥厚的发病机制非常复杂,涉及神经激素激活、代谢改变、细胞重编程和多种信号通路的调节。尽管人们在治疗心衰方面取得了重大进展,但从心脏肥厚到心力衰竭的机制仍然不明确。近日,云序生物助力哈尔滨医科大学杨宝峰院士团队在Advanced science期刊(IF=14.3)发表题为“The m7G Methyltransferase Mettl1 Drives Cardiac Hypertrophy by Regulating SRSF9-Mediated Splicing of NFATc4”的研究论文,该论文探究Mettl1的作用及其在非缺血性心肌肥厚和心衰中的作用机制,阐明了Mettl1介导的RNA的N7甲基鸟苷修饰可造成心脏肥厚,为HF提供了新的治疗靶点。本次研究中,云序生物提供m7G AlkAniline-Seq、m7G MeRIP-seq、RNA-seq及生信分析服务。本文特色,关于m7G修饰的检测,研究者同时采用了抗体富集的方法m7G MeRIP-seq以及单碱基分辨方法m7G AlkAniline-Seq,实现对mRNA上m7G位点的精确检测。

标题:The m7G Methyltransferase Mettl1 Drives Cardiac Hypertrophy by Regulating SRSF9-Mediated Splicing of NFATc4
发表时间:2024.8.7
发表期刊:Advanced science
样品类型:小鼠心肌细胞
研究方法:m7G AlkAniline-Seq、m7G MeRIP-seq、RNA-seq等
研究摘要
心脏肥厚是导致心力衰竭(HF)的关键因素,但其发病机制尚未完全阐明。Mettl1催化的RNA n7 -甲基鸟苷(m7G)修饰与缺血性心脏损伤和纤维化有关。本研究旨在阐明Mettl1的作用及其在非缺血性心肌肥厚和心衰中的作用机制。研究发现,在横断主动脉收缩(TAC)和血管紧张素II (Ang II)输注后,Mettl1在人类衰竭心脏和小鼠肥厚心脏中上调。YY1在心肌肥厚过程中作为Mettl1的转录因子。敲除Mettl1可减轻TAC或Ang II刺激所引起的心脏肥厚及其功能障碍。相反,心脏特异性的Mettl1过表达会导致心脏重塑。从机制上讲,Mettl1通过诱导SRSF9 mRNA的m7G修饰增加SRSF9的表达,促进NFATc4的选择性剪接和稳定,从而加速心脏肥厚。此外,在体内和体外,敲低SRSF9可防止TAC或Mettl1介导的心脏肥厚。该研究确定Mettl1是心脏肥厚的关键调节因子,为心力衰竭提供了新的治疗靶点。
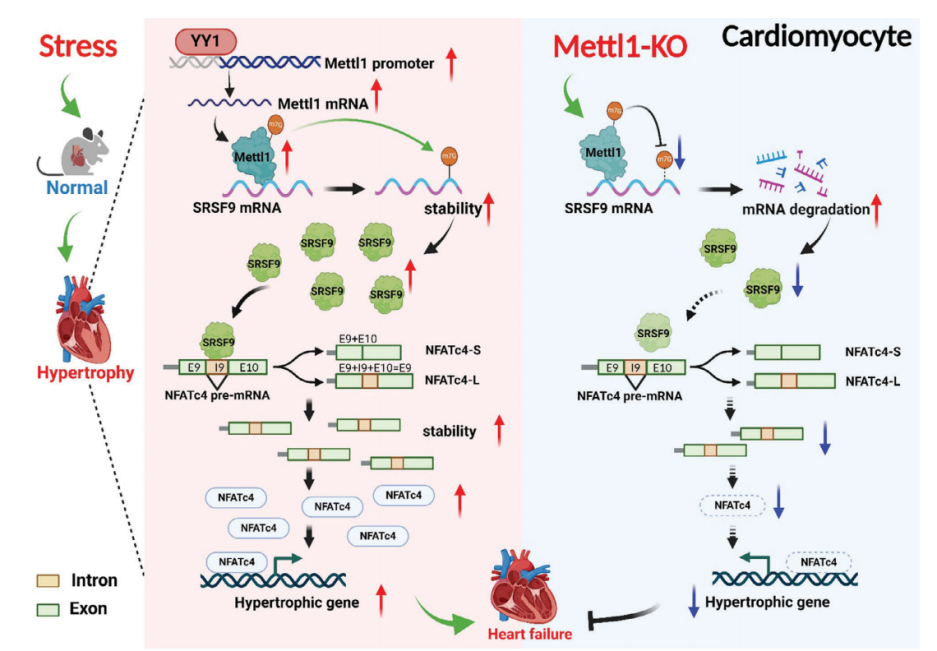
技术路线

研究结果
1.YY1诱导心肌肥厚过程中Mettl1表达上调
为了研究m7G甲基转移酶Mettl1是否参与心脏肥厚,我们评估了Mettl1在心力衰竭患者心脏中的表达。免疫印迹分析显示,与非衰竭对照组相比,衰竭心脏中Mettl1蛋白表达显著增加(图1A)。为了验证这一发现,我们建立了横断主动脉收缩(TAC)引起的心脏压力过载小鼠模型。如图1B所示,在肥厚心脏中,心肌肥厚基因表达上调,同时总RNA中m7G修饰水平升高。Western blot和qRT-PCR分析显示,与对照组相比,肥厚心脏中Mettl1 的mRNA和蛋白水平显著上调(图1C,D)。同样,与盐水处理的对照组相比,Ang II处理的小鼠心脏中Mettl1的mRNA和蛋白表达均升高(图1F,G)。此外,免疫荧光染色显示,在TAC上定位于心肌细胞,心肌蛋白Mettl1表达的增加,Mettl1与α-actinin共定位证明了这一点(图1E)。与体内结果一致,Ang II诱导的心肌细胞肥厚模型显示新生小鼠心肌细胞(NMCMs)中RNA m7G水平和Mettl1表达显著增加。这些发现提示Mettl1可能通过介导m7G修饰参与心脏肥厚过程。
随后,我们继续阐明心脏压力过载时Mettl1表达的上游机制。最近的一项研究发现反式转录因子1(SP1)和Yin Yang-1(YY1)是Mettl1的潜在转录因子。为了研究这一点,我们构建了YY1和SP1的siRNAs,并分别检测了敲除YY1或SP1后Mettl1表达的变化。在注入Ang II后的NMCMs中,YY1敲除后,Mettl1的mRNA和蛋白水平均显著降低(图1H,I)。然而,SP1的敲除对Mettl1的表达没有影响。这些结果提示YY1可能是Mettl1的一个转录因子。为了证实这一假设,我们构建了含有Mettl1启动子的荧光素酶载体,并将其转染到经Ang II处理的NMCMs中。荧光素酶分析结果显示,Ang II增加了Mettl1启动子活性,而沉默YY1后,这种活性显著降低(图1J)。此外,染色质免疫沉淀 (ChIP-qPCR)显示YY1与Mettl1启动子结合,在Ang II处理后相互作用增强(图1K)。综上所述,这些发现表明YY1在心肌肥大过程中作为上游调节因子激活Mettl1的表达。
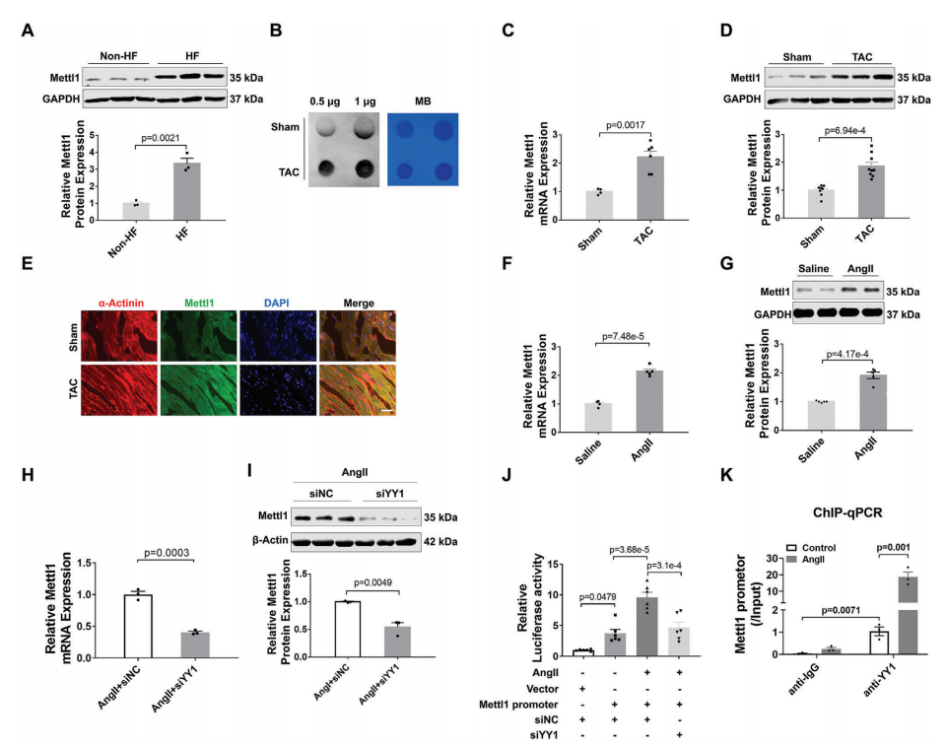
图1:在肥厚小鼠心脏中,YY1转录激活Mettl1。
(A)Western blotting分析心脏组织中Mettl1蛋白水平。
(B)Dot blot分析心脏组织中m7G修饰水平。
(C)qRT-PCR分析Mettl1 mRNA水平。
(D)Western blot分析心脏组织中Mettl1蛋白水平。
(E)α-actinin和Mettl1染色的代表性免疫荧光图像。
(F)心脏组织Mettl1 mRNA水平的qRT-PCR分析。
(G)免疫印迹分析Mettl1心脏组织中的蛋白质含量。
(H)qRT-PCR检测NMCMs中Mettl1 mRNA的表达。
(I)NMCMs Mettl1蛋白水平的变化。
(J)NMCMs后的荧光素酶活性变化。
(K)YY1或IgG抗体进行ChIP-qPCR分析。
2.缺乏Mettl1可减轻心脏压力过载引起的心肌肥厚和心衰
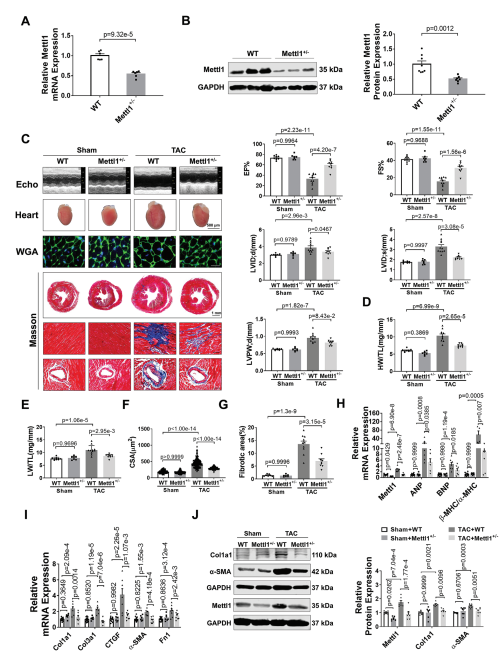
为了研究Mettl1在心脏肥厚中的功能作用,我们制造了全身Mettl1基因敲除小鼠。虽然Mettl1杂合子敲除(Mettl1 KO)是存活和可育的,但与野生型(WT)小鼠相比,Mettl1 KO小鼠心脏Mettl1 mRNA和蛋白水平显著降低(图2A,B)。Mettl1 KO和年龄匹配的WT幼崽接受TAC处理诱导心脏压力过载,导致血流速度增加,证实了与心脏压力过载相同的水平。Mettl1 KO和WT小鼠在体重、心脏重量和心功能方面没有显著差异(图2C)。然而,在TAC处理 10周后,Mettl1 KO小鼠的心脏重量与体重之比(HW/WB)、心脏重量与胫骨长度之比(HW/TL)和肺重量与胫骨长度之比(LW/TL)相对于WT小鼠有所下降(图2D,E)。超声心动图分析显示,与WT小鼠相比,TAC处理后Mettl1 KO小鼠的心功能得到改善,射血分数(EF%)和缩短分数(FS%)增加。此外,TAC诱导的WT小鼠收缩期左室内径(LVID;d)、舒张期左室内径(LVID;s)和舒张期左室后壁厚度(LVPW;d)的增加幅度在Mettl1 KO小鼠中有所减弱(图2C)。小麦胚芽凝集素(WGA)染色显示,与WT小鼠相比,TAC处理后Mettl1 KO小鼠的心肌细胞横截面积(CSA)显著减少(图2F)。同时,与TAC处理后的WT心脏相比,Mettl1 KO心脏中心脏胎儿基因ANP、BNP和β-MHC/α-MHC的mRNA表达下调(图2H)。此外,Mettl1 KO心脏在TAC处理后表现出明显的心脏血管周围和间质纤维化减少,纤维化基因Col1a1、Col3a1、CTGF、α-SMA和纤维连接蛋白1 (Fn1)的表达减少,Col1a1和α-SMA的蛋白水平也降低(图2G,I,J)。
为了进一步研究Mettl1在心脏肥厚中的作用,将Mettl1 KO小鼠和WT对照小鼠进行为期4周的Ang II输注(每天2.5 mg kg−1)。经Ang II治疗后,Mettl1 KO和WT小鼠的体重、血压或心率均无显著差异。Ang II诱导了Mettl1 mRNA和蛋白表达的显著增加,而在Mettl1 KO小鼠的心肌中,这种表达有所减弱。超声心动图显示,Ang II后Mettl1 KO小鼠心功能障碍得到改善,EF%和FS%增加,LVID;s和LVPW;d降低。此外,与WT小鼠相比,Mettl1 KO小鼠表现出更少的心脏肥厚,心脏大小、HW/TL、LW/TL、HW/BW和心肌细胞CSA在Ang II后均减小。与这些发现一致,在Ang II后,Mettl1 KO小鼠的心脏胎儿基因表达比WT对照组低。这些结果表明,Mettl1缺乏可以防止压力过载时心脏重构和HF的发生。
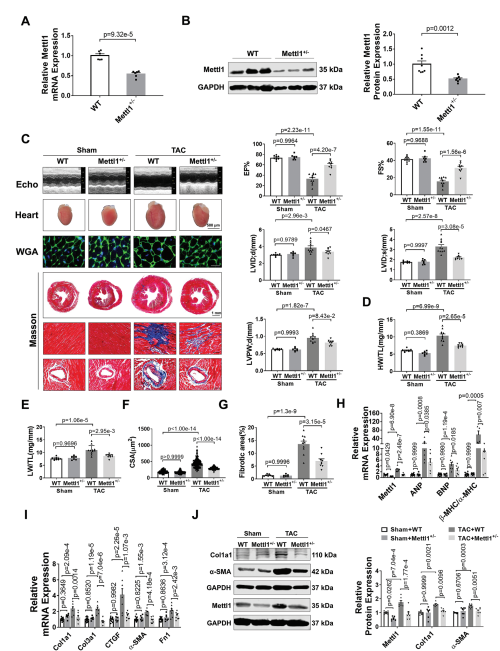
图2:缺乏Mettl1可减轻心脏压力过载后的心肌肥厚和心衰。
(A)qRT-PCR分析心脏组织中Mettl1 mRNA表达的qRT-PCR分析。
(B)Western blot分析心脏组织中Mettl1蛋白水平。
(C)超声心动图测量的原始痕迹。
(D)sham和TAC处理小鼠心脏重量(HW)与胫骨长度(TL)的相对 比值。
(E)sham和TAC处理小鼠的相对肺重(LW)与胫骨长度(TL)之比。
(F)心室心肌细胞横截面积(CSA)的量化。
(G)sham和TAC组小鼠Masson染色。
(H-I)qRT-PCR分析小鼠基因变化。
(J)免疫印迹分析心脏组织蛋白质含量。
3.Mettl1驱动心脏重塑
上述结果提示进一步研究Mettl1的过表达是否可以复制在压力过载诱导的心脏重构中观察到的表型。我们采用了一种功能获得策略,使用腺相关病毒血清型9编码Mettl1转录物(AAV9-Mettl1),以AAV9载体作为阴性对照(AAV9-Null)。与AAV9载体处理的小鼠相比,AAV9-Mettl1处理导致Mettl1 mRNA和蛋白水平显著上调,达到与TAC处理的心脏相似的水平(图3A,B和1C,D)。值得注意的是,过表达Mettl1显著降低EF%和FS%,引起心脏扩张(图3C)。Mettl1过表达小鼠的心肌肥厚也很明显,LVPW;d、HW/TL和CSA的显著增加证明了这一点(图3C-E)。此外,我们评估了从AAV9-Null和AAV9 - Mettl1处理的小鼠中分离的成年小鼠心肌细胞的表面积。与AAV9 - null处理的小鼠相比,Mettl1过表达的心肌细胞表面积一致增加。同样,Mettl1过表达导致离体心肌细胞的宽度明显增加,长度适度增加。Mettl1过表达后心脏胎儿基因mRNA表达的显著升高进一步支持了这一点(图3F)。此外,Masson染色显示,Mettl1过表达诱导心脏纤维化,纤维化基因mRNA表达显著增加,Col1a1和α-SMA蛋白水平升高(图3G-I)。
接下来,我们进行了体外研究,探讨Mettl1在心肌细胞肥大中的作用。为此,在Ang II治疗之前,将靶向Mettl1的siRNA转染到NMCM中。siMettl1处理导致NMCM中Mettl1蛋白水平显著降低。免疫荧光染色检测到,通过敲低Mettl1可以阻止Ang II诱导的细胞增大。同样,Mettl1的敲低减弱了Ang II诱导的ANP、BNP和β-MHC mRNA表达的上调。相反,用含有Mettl1基因的腺病毒(Adv)感染NMCM,人工过表达Mettl1,会产生相反的结果。具体来说,Mettl1过表达导致心肌细胞肥大,心肌细胞大小、ANP和BNP mRNA水平增加。这些结果共同表明,Mettl1足以诱导心脏重构。
为了阐明Mettl1驱动心肌肥厚的机制,我们采用RNA-seq结合m7G MeRIP-seq,分别在Mettl1过表达的小鼠心肌和NMCM中进行全转录组分析和m7G位点定位。Mettl1过表达后,NMCM中总m7G甲基化水平更高,而Mettl1敲低则具有相反的效果。m7G MeRIP-seq分析显示,在过表达Mettl11的心脏中,有3085个上调基因和5372个高甲基化峰。此外,m7G峰值水平与基因表达之间的相关性表明,1520个上调基因的m7G修饰增加。在综合m7G MeRIP-seq和m7G AlkAnilineSeq结果后,我们发现566个重叠基因在过表达Mettl1后,m7G峰和mRNA表达增加(图4A)。KEGG分析表明,566个重叠基因主要富集于与心脏肥厚相关的信号通路,如AMPK、MAPK和mTOR通路。对566个重叠基因GO分析显示,参与的生物过程与心脏发育、血管生成和DNA损伤有关。此外,一些基因表现出蛋白质和核酸的结合能力。为了确定Mettl1的具体靶点,我们综合参考了KEGG和GO分析的结果,得出了14个可能参与心脏肥大相关途径的基因(图4B)。然后,我们评估了这些候选基因在TAC处理和过表达Mettl1的心脏中的表达,发现14个基因中有4个在两种情况下都表现出上调。此外,m7G MeRIP-qPCR结果显示,与抗IgG相比,抗m7G显著免疫沉淀m7G甲基化的SRSF9 mRNA在NMCM和小鼠心肌中,Mettl1过表达显著增强了这种富集(图4C)。相反,TAC诱导的SRSF9 mRNA m7G水平升高在Mettl1缺陷心脏中减弱。整合基因组分析显示,Mettl1过表达后,NMCM中m7G水平和SRSF9 mRNA表达显著增加(图4D)。为了评估Mettl1是否会影响SRSF9 mRNA的稳定性,我们在NMCM中进行了RNA衰减试验。结果显示,在放线菌素D处理后,Mettl1的敲低加速了SRSF9 mRNA的衰减,而Mettl1的过表达阻止了SRSF9 mRNA在心肌细胞中的衰减(图4E,F)。无论是否进行TAC处理,Mettl1 KO心脏的SRSF9蛋白表达均显著降低(图4G)。相反,Mettl1过表达显著升高SRSF9蛋白水平(图4H)。在体外NMCM中也观察到类似的结果(图4I,J)。
m7G AlkAnilineSeq分析显示,两个m7G峰位于SRSF9 mRNA的3'UTR上。此外,我们通过catRAPID数据库预测了Mettl1对SRSF9 mRNA的结合偏好,结果显示Mettl1对SRSF9 mRNA的3'UTR区域具有更强的结合偏好。为了进一步证实SRSF9是Mettl1的直接靶点,我们构建了一个包含野生型SRSF9编码序列(CDS)和3'UTR区域(SRSF9- wt)的质粒,以及一个突变型SRSF9 3'UTR (SRSF9- mut)质粒(图4K)。Western blotting证实两种质粒在NMCM中均显著过表达SRSF9。正如预期的那样,m7GMeRIP-qPCR结果显示SRSF9-WT能够被抗m7G免疫沉淀,而SRSF9-Mut则不能(图4L)。此外,Mettl1促进了转染的外源SRSF9转录物在SRSF9- wt处理的心肌细胞中的稳定性,而这种作用在SRSF9- mutt处理的心肌细胞中被消除(图4M)。这些结果表明Mettl1以m7G依赖性的方式调控SRSF9的表达。
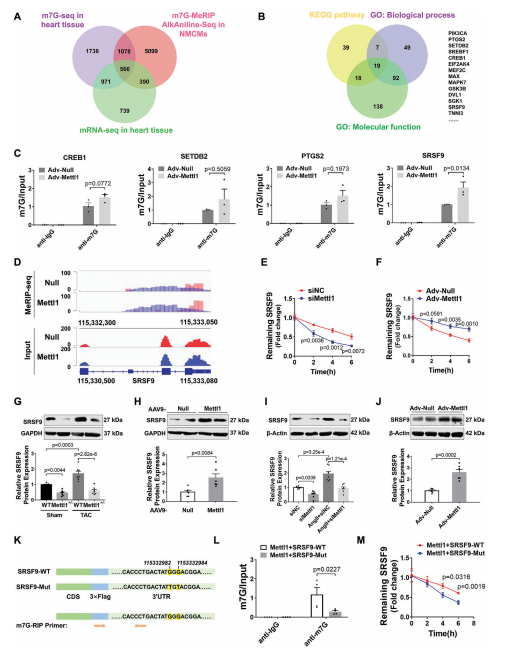
5.缺乏SRSF9可抑制心脏重构
为了研究SRSF9在心肌细胞肥大中的功能,我们设计了三种不同的siRNA序列来沉默NMCM中的SRSF9。结果显示,用siSRSF9 2#处理后,SRSF9蛋白表达下调最为显著。SRSF9的敲低减弱了Ang II诱导的心肌细胞大小和肥厚基因表达的增加,而SRSF9的过度表达则显著增加了心肌细胞肥厚。为了证实我们的体外实验结果,我们进行了一项体内功能丧失研究,以探索SRSF9在心脏肥厚中的作用。为此,我们构建了一个携带SRSF9- shRNA片段的AAV9 (AAV9- shSRSF9),在小鼠心肌中敲除SRSF9。小鼠接受相同水平的TAC处理,主动脉弓血流量增加。与AAV9-shSRSF9处理的小鼠相比,给予AAV9-shSRSF9可显著降低SRSF9 mRNA和蛋白水平,并显著消除TAC诱导的SRSF9表达升高(图5A,B)。超声心动图分析显示,敲低SRSF9可显著缓解TAC诱导的心功能障碍,降低心脏扩张,降低LVPW;d(图5C)。此外,SRSF9的敲除减弱了HW/TL和LW/TL的增加(图5D,E)。WGA染色显示,TAC诱导CSA显著增加,沉默SRSF9后CSA增加被消除(图5F)。与此一致的是,TAC诱导的心脏肥厚基因表达水平的增加可以通过敲低SRSF9消除(图5H)。此外,抑制SRSF9可降低TAC后心脏纤维化和纤维化基因的表达(图5G,I)。这些结果表明,阻断SRSF9可减轻压力过载时的心肌肥厚。
为了研究SRSF9过表达是否会导致与压力过载引起的心脏肥厚相同的表型,我们采用了功能获得法,通过感染AAV9-SRSF9及其阴性对照AAV9-Null。与AAV9载体处理的小鼠相比,AAV9-SRSF9处理导致SRSF9 mRNA和蛋白水平显著上调。值得注意的是,SRSF9过表达显著降低EF%和FS%,并引起心脏扩张。在SRSF9过表达小鼠中也观察到心脏肥厚,表现为LVPW;d、HW/TL和CSA的显著增加。结果进一步验证了这一点,过表达SRSF9导致心脏胎儿基因表达显著增加。
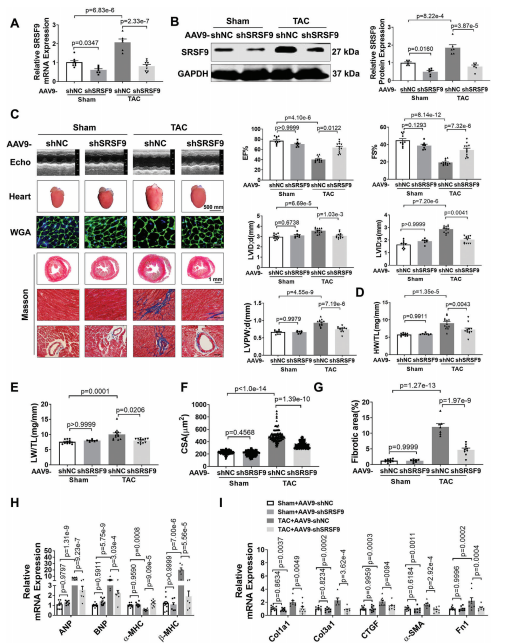
6.SRSF9通过调节剪接稳定NFATc4 mRNA
SRSF9是一种RNA结合蛋白,也参与RNA剪接。为了揭示SRSF9介导心脏肥厚的机制,我们基于ENCORI、catRAPID和KnockTF2.0三个数据库预测了可能与SRSF9结合的基因。如图6A所示,这些数据库的交集产生了已知与心脏肥厚有关的基因,包括EZH2、NFATc4、GATA4和JARID2。为了验证这些基因是否是SRSF9的靶标,我们在敲除SRSF9后测定了Ang II处理的NMCM中它们的mRNA表达。结果显示,Ang II使NFATc4 mRNA的表达升高最为显著,而SRSF9的敲低则显著降低了NFATc4 mRNA的表达(图6B)。相比之下,SRSF9过表达组的NFATc4 mRNA水平高于adv - null处理组(图6C)。Western blot结果显示,敲低SRSF9消除了Ang II诱导的NFATc4蛋白水平的升高,而过表达SRSF9则增加了NFATc4的表达(图6D,E)。同样,通过沉默SRSF9,TAC诱导的小鼠心脏中NFATc4 mRNA和蛋白水平上调被减弱,而心脏组织中SRSF9的过表达导致NFATc4表达显著上调。NFATc4已被证明通过易位进入细胞核促进胎儿基因的转录,从而促进心脏肥厚。因此,我们在处理SRSF9后检测了NFATc4在细胞核和细胞质中的分布。正如预期的那样,Ang II导致NFATc4的核积累,而SRSF9的敲低减轻了这种积累(图6F)。相比之下,SRSF9过表达导致细胞核部分NFATc4增加(图6G)。
鉴于SRSF9是一个剪接因子,我们推断SRSF9可能调控NFATc4的选择性剪接。为了验证这一假设,我们采用琼脂糖凝胶电泳检测了之前发现的两个NFATc4转录体变体的剪接率(PSI)。结果显示,敲低SRSF9可降低NFATc4长变体转录本(NFATc4- l)的PSI,提示SRSF9可能以一种内含子保留的方式促进NFATc4的剪接(图6H)。根据RBPmap数据库,我们在NFATc4的第9和第10外显子之间的第9内含子上预测了潜在的SRSF9结合位点(AGGAGAA和GGATGGA序列),用红色字体标记(图6I)。这促使我们验证SRSF9是否通过直接结合其pre-mRNA来调节NFATc4剪接。为此,我们构建了一个包含NFATc4 9 - 10外显子基因组DNA片段和内含子9的WT小基因报告质粒(NFATc4-WT)和一个在NFATc4结合位点突变的突变小基因报告质粒(NFATc4- mut)(图6J)。与转染的外源NFATc4- wt组相比,SRSF9结合位点的突变导致NMCM中SRSF9刺激的NFATc4内含子保留事件的富集减少,表明SRSF9以序列特异性的方式调节NFATc4剪接(图6J)。此外,RIP-qPCR实验表明外源NFATc4-WT可以与SRSF9结合,而外源NFATc4-Mut则失去了这种能力(图6K)。
为了进一步研究SRSF9对NFATc4转录变体mRNA水平上的影响,我们设计了两对引物,分别特异性扩增NFATc4-L或NFATc4的较短转录变体(NFATc4- s)。qRT-PCR结果显示,基线时NMCM中NFATc4-L的mRNA表达水平远高于NFATc4 pre-mRNA和NFATc4-S。值得注意的是,在TAC诱导的肥厚心脏中,NFATc4-L的表达显著上调,而SRSF9的沉默则减弱了NFATc4-L的表达。SRSF9过表达增加,而SRSF9敲低降低了NFATc4-L的mRNA表达(图6L,M)。此外,我们检测了NFATc4-L和NFATc4-S的mRNA衰减率,发现NFATc4-L的稳定性比NFATc4-S强得多,并且SRSF9的敲低加速了NFATc4-L的降解。为了进一步阐明SRSF9的哪个结构域负责调节NFATc4 pre-mRNA剪接,我们分别通过去除RNA识别结构域1和2 (Flag-SRSF9-ΔRRM1和Flag-SRSF9-ΔRRM2)构建了两个标记为SRSF9的flag标记截断。如图6N所示,SRSF9 RRM1结构域的缺失而非RRM2结构域的缺失显著抑制了NFATc4的选择性剪接。此外,当SRSF9在其RRM1结构域缺失时,SRSF9与NFATc4pre-mRNA的结合能力降低(图6O)。进一步的实验表明,RRM1结构域的缺失导致SRSF9丧失了调控NFATc4-L mRNA和NFATc4蛋白表达的能力(6P, 6Q)。综上所述,我们的数据表明SRSF9通过调节NFATc4 pre-mRNA的剪接来增加NFATc4蛋白的表达。
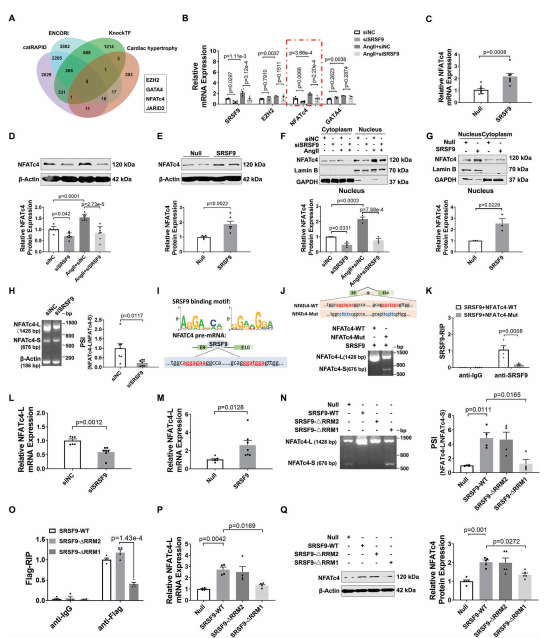
7.SRSF9介导Mettl1诱导的心肌肥厚
接下来,我们研究了Mettl1诱导的心肌肥厚是否由SRSF9介导。为了解决这个问题,我们进行了一项救援实验,通过AAV9-shSRSF9治疗小鼠,在Mettl1过表达的心脏中敲除SRSF9。正如预期的那样,相对于AAV - shNC处理组,AAV9-shSRSF9处理显著抑制了Mettl1过表达引起的SRSF9表达的增加(图7A,B)。引人注目的是,Mettl1诱导的心功能障碍在很大程度上通过敲低SRSF9得以恢复(图7C)。敲低SRSF9可阻止过表达Mettl1的心脏中HW/TL的增加(图7D)。此外,通过沉默SRSF9,心肌肥厚(CSA增加,Mettl1过表达导致肥厚基因表达)得到减弱(图7E,G)。此外,敲低SRSF9可减轻Mettl1诱导的心脏纤维化(图7F,H)。肥厚心脏和心肌细胞中的NFATc4蛋白水平显著升高,而Mettl1的敲低则消除了NFATc4蛋白水平。此外,Mettl1的敲低抑制了NFATc4-L的PSI和NFATc4 mRNA的稳定性,而Mettl1的强化表达在NMCMs中具有相反的结果。为了证实体内结果,通过操纵NMCMs中Mettl1和SRSF9的表达,进行了一项功能获得和功能丧失研究。Mettl1引起心肌细胞大小和胎儿基因表达的显著增加,可以通过敲低SRSF9来恢复。此外,通过沉默SRSF9,可以减少Mettl1过表达导致的NFATc4蛋白表达的显著增加(图7I)。相反,SRSF9的过表达抵消了Mettl1敲低介导的心肌细胞肥厚表型的减弱,这反映在心肌细胞大小、胎儿基因表达和NMCMs中NFATc4蛋白表达的增加上。
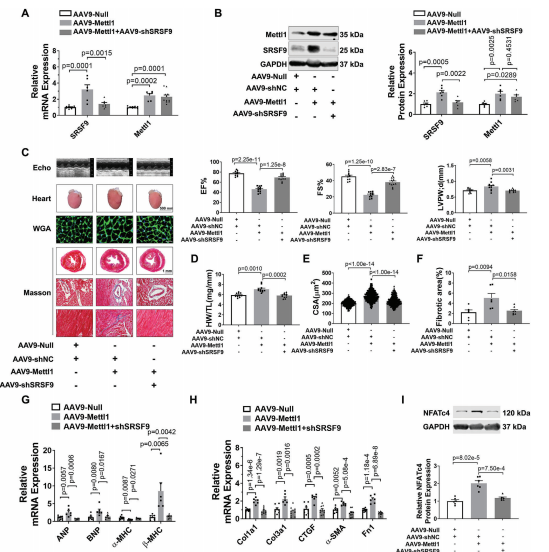
全文小节
· Mettl1在人类衰竭心脏和小鼠肥厚心脏中表达升高,与RNA中m7G水平升高相关。
· YY1在心肌肥厚过程中作为Mettl1的转录激活因子。
· Mettl1在压力过载条件下驱动心脏肥大和重塑中起关键作用。
· 缺乏SRSF9对TAC手术具有心脏保护作用,可挽救mettl1诱导的心肌肥厚。
· Mettl1诱导SRSF9 mRNA的m7G修饰,增加SRSF9的稳定性,进而与NFATc4结合并剪接,导致促肥厚表型。
· 本文特色,关于m7G修饰的检测,研究者同时采用了抗体富集的方法m7G MeRIP-seq以及单碱基分辨方法m7G AlkAniline-Seq,实现对mRNA上m7G位点的精确检测。
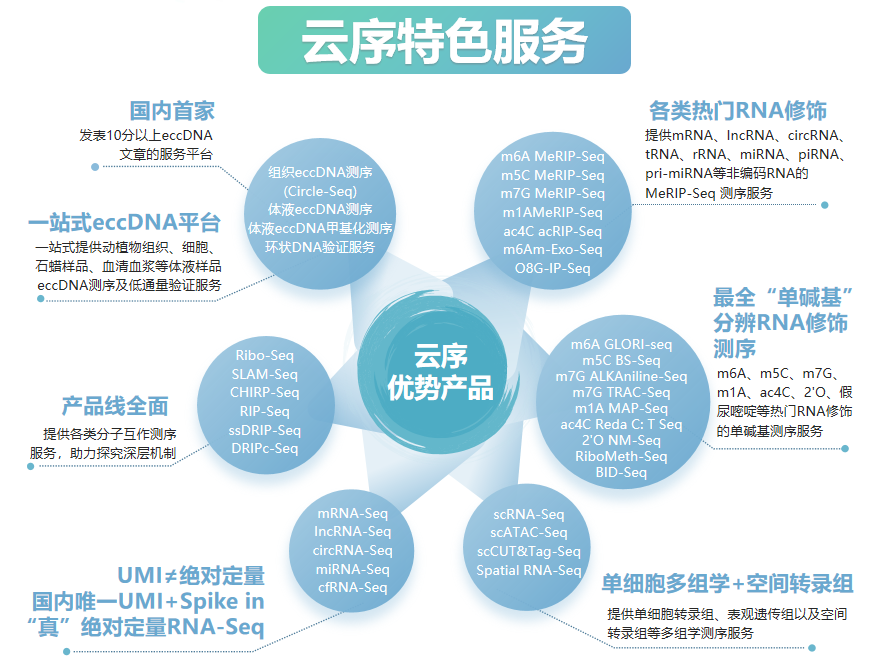
云序生物RNA修饰优势
优势一:RNA修饰产品线最全,热门修饰IP类/单碱基分辨测序 + IP试剂盒
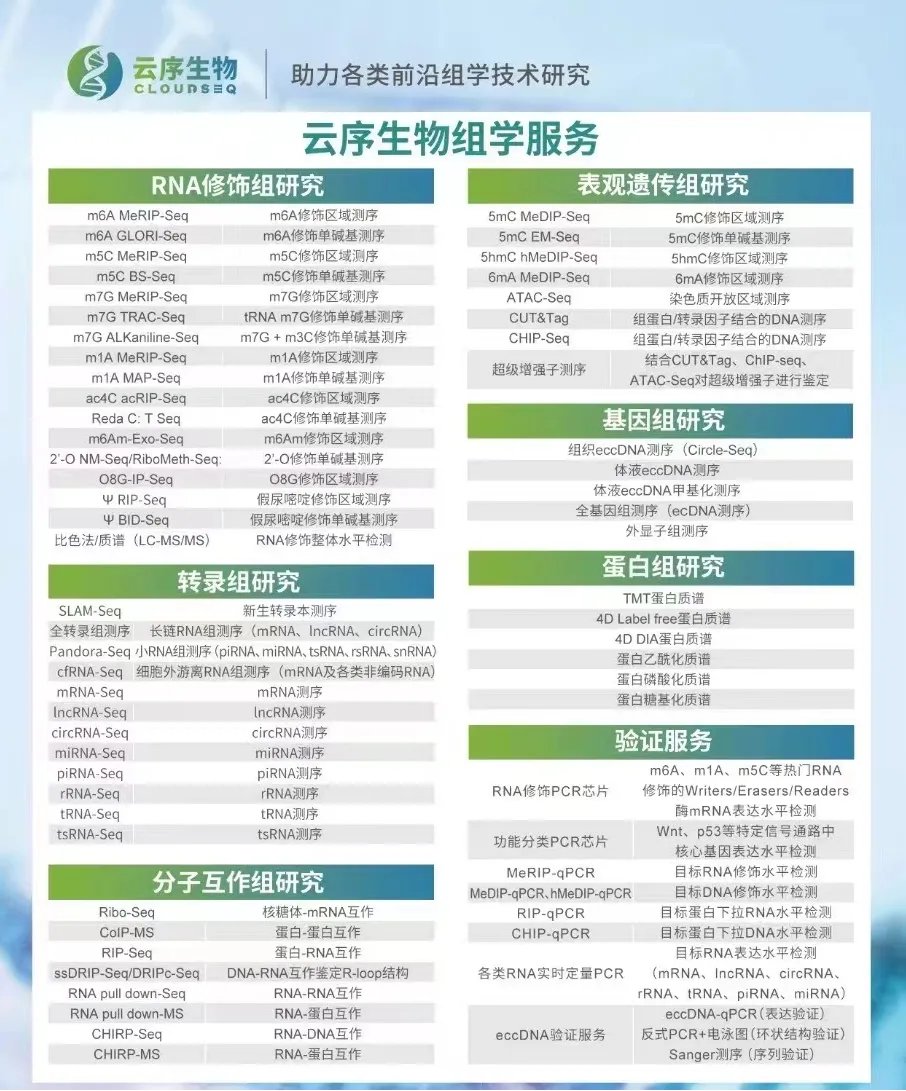
RNA修饰测序是云序生物转录调控及表观组学旗下的重磅产品。在国内,目前云序生物具有最全的RNA修饰测序平台,可提供m6A、m5C、m1A、m7G、m3C、O8G、m6Am、ac4C乙酰化、2'-O-甲基化和假尿嘧啶等各类热门修饰的多种测序技术,包括IP类和单碱基分辨测序,涵盖各类RNA分子,如mRNA、lncRNA、circRNA、tRNA、rRNA、miRNA及pri-miRNA等非编码RNA的MeRIP-seq服务。通过不断迭代优化,目前RNA修饰测序平台适用于各类物种,组织及RNA起始量的个性化需求。同时,云序生物还在线出售m6A、m5C、m1A、m7G、O8G、ac4C的IP试剂盒。
云序RNA修饰测序产品列表:
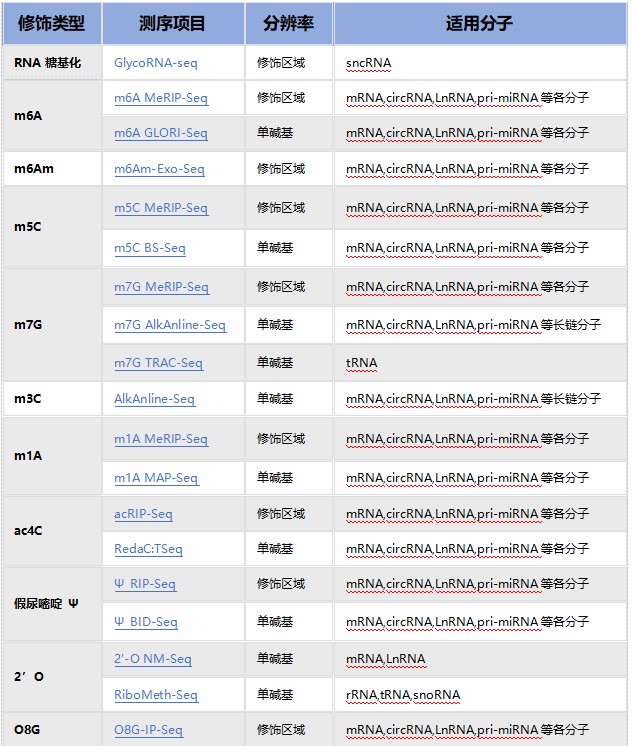
云序RNA修饰试剂盒产品列表:
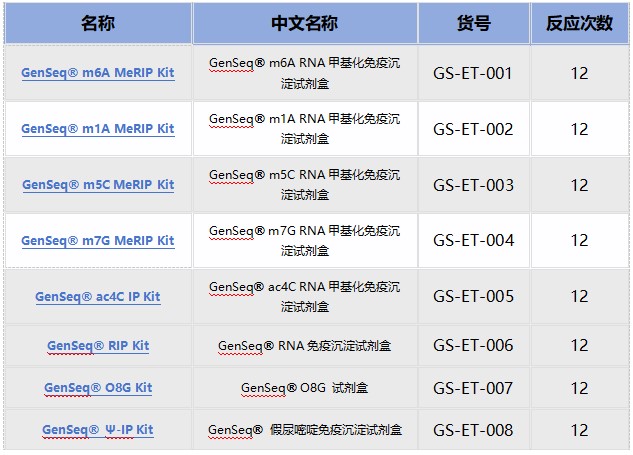
优势二:数千篇SCI论文、数万例测序样本
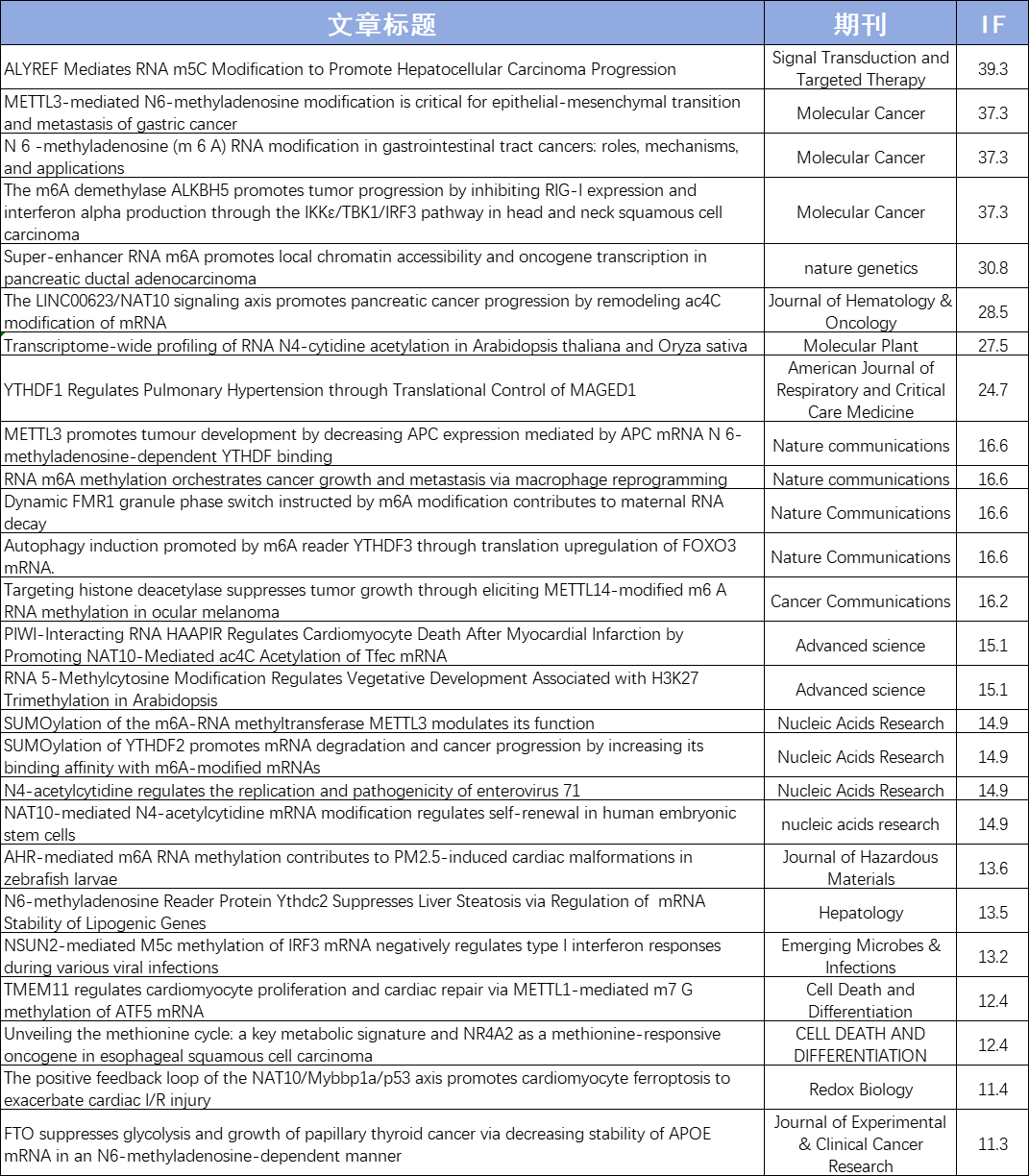
目前,云序累计助力客户发表160+篇RNA修饰SCI论文,影响因子1000+,累计完成数千例RNA甲基化测序样本,全面覆盖医口、农口等各类样本。文章涉及多种组学联合方案,如RNA修饰联合RNA-seq、Ribo-seq、RIP-seq、SLAM-seq及单细胞等。云序生物已成为客户心中值得信赖的RNA修饰合作伙伴!
云序客户RNA修饰部分文章列表:
优势三:MeRIP-seq全面升级,spike in严格质控IP实验
云序m6A meRIP-seq Plus(spike in)通过在实验过程中加入两类人工合成的spike in:阳性Spike-In(带m6A修饰)和阴性Spike-In(不带修饰),与样品一起进行MeRIP实验及后续测序分析,根据spike in的检测信号对MeRIP实验进行质控,确保实验流程的准确性、可重复性及抗体的特异性!

优势四:测序到验证、靶标定位到分子机制,云序提供一站式服务
RNA修饰测序是云序生物转录调控及表观组学旗下的重磅产品。在国内,目前云序生物具有最全的RNA修饰测序平台,可提供m6A、m5C、m1A、m7G、m3C、O8G、m6Am、ac4C乙酰化、2'-O-甲基化和假尿嘧啶等各类热门修饰的多种测序技术,包括IP类和单碱基分辨测序,涵盖各类RNA分子,如mRNA、lncRNA、circRNA、tRNA、rRNA、miRNA及pri-miRNA等非编码RNA的MeRIP-seq服务。通过不断迭代优化,目前RNA修饰测序平台适用于各类物种,组织及RNA起始量的个性化需求。同时,云序生物还在线出售m6A、m5C、m1A、m7G、O8G、ac4C的IP试剂盒。
云序多组学方案:
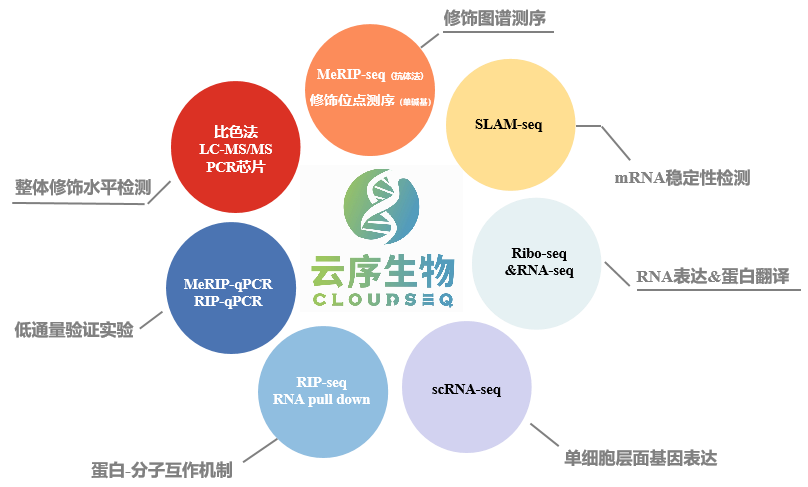
RNA修饰多组学已经广泛应用于各类研究中,除了常见的物种以外,在很多非模式生物上也有研究,如牛羊、酵母、细菌、番茄、玉米等等。研究思路也多种多样,如果您有更多的想法及需求,欢迎后台与我们联系!
往期回顾
黄荷凤院士团队前沿成果 | 云序m6A MeRIP-Seq&RIP-seq技术助力揭示高龄父亲引起子代神经炎症风险加剧
聚焦表观,转录共荣!墨卓生物与云序生物达成合作
PNAS/Q1 云序生物助力苏大陶金团队揭示m6A介导神经病理性疼痛新机制
IF=20.4 高分ac4C新鲜出炉||云序助力客户探究ac4C与肿瘤和免疫相关新机制
Get新热点! m6A&单细胞强强联合,助力文章升级! | m6A&单细胞专题一
云序客户近期RNA修饰高分文章速览
Nature Genetics IF=30.8|非编码seRNA m6A修饰介导组蛋白修饰和癌基因表达
云序客户| m6A MeRIP-seq助力揭示早发糖尿病表观调控新机制
云序客户m6A高分文章|揭示组蛋白乙酰化与m6A修饰在眼部黑色素瘤发生中的共同作用机制

下期精彩继续